In this study we found that the complement inhibition by OSCS was also dependent on C1inh, as inhibition by OSCS disappeared with the depletion of C1inh from plasma. Surface plasmon resonance assay showed OSCS increased the binding of C1 inhibitor with C1s protease, the initial component of the classical complement pathway. Therefore OSCS inhibits the complement classical pathway by potentiating the interaction of C1inh with C1s. The GAG enhancement of the C1inh – C1s interaction as a direct cause of complement inhibition is consistent with the crystal structure of C1inh that was determined for the serpin domain of recombinant C1inh in its latent form. Based on this structure, surface charge pattern, heparin affinity measurements, and docking of a heparin disaccharide, a heparin binding site is proposed in the contact area of the serpinproteinase encounter complex. Beinrohr et al proposed that by binding to C1inh and neutralizing its positively charged surface patches the polyanions facilitate the C1inh-C1s interaction. This can explain how the inhibitory activity of C1 inhibitor toward proteases, such as C1s or activated factor XI, can be greatly enhanced by heparin and other glycosaminoglycans. Our data support this model for the enhancement of C1inh C1s interaction by GAGs that we observed in the current study, as well as in our earlier work. Heparin lots contaminated with OSCS can inhibit complement activity in vitro. However the sustained levels of 10�C20 microgram/ mL are unlikely with intravenous dosing of heparin although subcutaneous administration of contaminated heparin may have allowed for higher local levels of OSCS. A veterinary drug, polysulfated glycosaminoglycan that is very similar in structure to OSCS is still used in animals and administered locally. PSGAG is also a polysulfated chondroitin sulfate with 3 to 4 sulfate groups per disaccharide unit and is considered to be a disease-modifying veterinary drug for osteoarthritis. PSGAG is anti-inflammatory and many mechanisms have been postulated from preservation of joint glycosaminoglycans to inhibition of PGE2 synthesis, toxic oxygen radical generation, and complement activation. Studies have shown an impact of PSGAG at relatively higher doses on complementmediated lysis of red blood cells without a clear mechanism of action. Our in vitro experiments using bacteria as model indicate PSGAG is a very strong inhibitor of complement fixation of bacteria. The potentiation of C1inh interaction with C1s by OSCS can also explain the effect of PSGAG on complement lysis and provide a mechanism for studies suggesting an increased likelihood of Compound Library inhibitor infections with intra-articular injection of PSGAG and low levels of bacteria. Although there was an increase in the absolute numbers of infections reported during the 2007�C2008 timeframe of the OSCS contamination, the relative numbers decreased. It may be of value to further assess GAG related products including PSGAG, for infection related adverse events, although adverse event reporting has many limitations. Based on the concentrations heeded to inhibit complement, there may not be an in vivo effect unless ![]() high doses are administered locally rather than systemically. There have been suggestions that glycosaminoglycans can be used to inhibit the complement activity in situations such as GANT61 autoimmune diseases.
high doses are administered locally rather than systemically. There have been suggestions that glycosaminoglycans can be used to inhibit the complement activity in situations such as GANT61 autoimmune diseases.
Month: August 2019
The observation that many CK2 substrates are proteins involved in cell survival
It is involved in several cellular processes, such as cell cycle, gene expression, protein synthesis, signal transduction and metabolism; however, its hall-mark is considered its prosurvival and anti-apoptotic function. This is supported by the reduction of CK2 activity or expression is invariantly followed by cell death, mainly due to apoptosis. Consistent with the anti-apoptotic function of CK2, cancer cells, which are characterized by rapid proliferation and defective apoptosis, express particularly high levels of CK2. It has a special role in tumorigenesis, potentiating pathways that are frequently up-regulated or untimely activated in cancer, and it has consequently been defined as “a key player in cancer biology”. Whenever comparison has been performed, CK2 has been shown significantly more abundant in tumor cells than in healthy counterparts. However, at the same time tumors rely more on CK2 for their survival, and this phenomenon, described as “addiction” to CK2 of cancer cells, explains why they are more SCH727965 msds sensitive to its inhibition or knocking-down, compared to normal cells. On these bases, CK2 is presently considered a promising therapeutic target, also exploiting the fact that, due to the peculiar structure of the CK2 catalytic site, several very specific 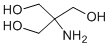 inhibitors are available. Many of them have already proved to be able to kill cancer cells and in some cases also employed for successful animal treatment. Apoptosis resistance is a major reason of cancer therapy failure; its mechanisms can be different and multifaceted, and is only partially understood. In many cases it is due to the expression of extrusion pumps of the ABC-transporter family, such as Pgp, which drive drugs outside the cell and reduce their effective concentration. Cells expressing these pumps are selected for their survival in response to treatment with a certain drug, but usually a cross-resistance occurs towards other compounds, even not structurally related; in these cases, cells are indicated as multidrug-resistant. Many other mechanisms have been reported to be involved in apoptosis resistance, including alteration in genetic INCB18424 JAK inhibitor features, DNA repair, drug target molecules, metabolic and growth pathways. In some cases, specific resistance is observed, such as that towards Imatinib and its derivatives targeting Bcr-Abl tyrosine kinase, frequently due to kinase mutations, but also to epigenetic changes, alternative splicing or induction of compensatory signaling pathways. CK2 has been already associated to the phenomenon of drug resistance: it phosphorylates Pgp and another extrusion pump, MRP1 and its inhibition allows a higher accumulation of drugs in Pgp or MRP1 expressing cells, suggesting that CK2 can up-regulate the Pgp function. Moreover, we have previously found that the CK2 catalytic subunit is overexpressed in a MDR cell line compared to the non-MDR counterpart, and that its overexpression contributes to the maintenance of the resistant phenotype. Here we evaluate the efficacy of the CK2 inhibitors CX-4945 and CX-5011 in a number of different cell lines, available as pairs, each pair containing a variant selected for resistance to druginduced apoptosis, and we demonstrate that these compounds can overcome the problem of drug resistance. We first measured CK2 activity in cells treated for different times with increasing concentrations of the compounds.
inhibitors are available. Many of them have already proved to be able to kill cancer cells and in some cases also employed for successful animal treatment. Apoptosis resistance is a major reason of cancer therapy failure; its mechanisms can be different and multifaceted, and is only partially understood. In many cases it is due to the expression of extrusion pumps of the ABC-transporter family, such as Pgp, which drive drugs outside the cell and reduce their effective concentration. Cells expressing these pumps are selected for their survival in response to treatment with a certain drug, but usually a cross-resistance occurs towards other compounds, even not structurally related; in these cases, cells are indicated as multidrug-resistant. Many other mechanisms have been reported to be involved in apoptosis resistance, including alteration in genetic INCB18424 JAK inhibitor features, DNA repair, drug target molecules, metabolic and growth pathways. In some cases, specific resistance is observed, such as that towards Imatinib and its derivatives targeting Bcr-Abl tyrosine kinase, frequently due to kinase mutations, but also to epigenetic changes, alternative splicing or induction of compensatory signaling pathways. CK2 has been already associated to the phenomenon of drug resistance: it phosphorylates Pgp and another extrusion pump, MRP1 and its inhibition allows a higher accumulation of drugs in Pgp or MRP1 expressing cells, suggesting that CK2 can up-regulate the Pgp function. Moreover, we have previously found that the CK2 catalytic subunit is overexpressed in a MDR cell line compared to the non-MDR counterpart, and that its overexpression contributes to the maintenance of the resistant phenotype. Here we evaluate the efficacy of the CK2 inhibitors CX-4945 and CX-5011 in a number of different cell lines, available as pairs, each pair containing a variant selected for resistance to druginduced apoptosis, and we demonstrate that these compounds can overcome the problem of drug resistance. We first measured CK2 activity in cells treated for different times with increasing concentrations of the compounds.
For C1s activity and an ELISA to test the inhibition of complement C4 and C3 deposition on immobilized aggregated
Reported that GAG family members, including R428 dextran sulfates with average heparin, heparan sulfate and CSA at concentrations from 100 to 1000 mg/ ml could inhibit complement. Dextran sulfate with an average MW of 500,000 had the strongest inhibition. The inhibition was due to GAG enhancement of the second-order rate constant of the inactivation of C1s by C1inh. OSCS was initially identified as a contaminant in certain lots of heparin that were associated with severe adverse events. Heparin is a polydisperse mixture of linear acidic polysaccharides, which is isolated by extraction from animal tissues, most commonly porcine intestines, and is a member of the glycosaminoglycan family. OSCS had been previously prepared from chondroitin sulfate, another member of the GAG family having similar backbone structure with heparin, by chemical sulfonation, and was shown to have anticoagulant activity. Patients that received the OSCS contaminated heparin developed hypotension, shortness of breath and GI symptoms compatible with contact system activation. In vitro studies showed OSCS activated contact system Factor XII and induced kinin-kallikrein activation. OSCS also induced the generation of the anaphylactoid toxins C3a and C5a in a manner that bypassed the C3 and C5 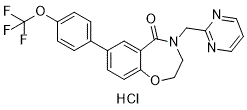 convertases but was also dependent on FXII. The impact of GAGs on complement and the observed effect of OSCS on complement components C3 and C5 suggested further evaluation of OSCS and complement. Investigation of the OSCS interactions with complement components using surface plasmon resonance by Linhardt��s group suggested that OSCS can bind to the complement components with moderate to high affinity comparable to that of heparin. Therefore a difference in the impact of OSCS-contaminated versus uncontaminated heparin is not explained by differences in binding to complement components. The impact of OSCS on the functional complement activity was not performed yet, which became the aim of our study. In earlier work, the complement function has been tested in vitro using a variety of models such as the standard 50% hemolytic complement assay, the enzyme immunoassay and the liposome immunoassay. In this study, we used an established model of Foretinib natural antibody mediated bacterial lysis through the complement classical pathway. The murine monoclonal polyreactive antibody 2E4 can bind with bacteria E. coli BL21, fix complement, lyse bacteria and generate anaphylatoxin C5a. This is a relevant model to study the impact of OSCS, OSCS-contaminated heparin lots, un-contaminated heparin lots and other GAGs on the complement classical pathway. Using this biologically relevant model, antibody mediated complement-dependant bacterial lysis, we demonstrated that OSCS can inhibit the complement classical pathway as indicated by lower level of C3 fixation on bacteria as well as decreased bacterial lysis. Initially, we hypothesized that C3 consumption might explain the decreased fixation of C3b on the antibodytreated bacteria. This hypothesis was consistent with the FXIIdependent OSCS induction of C3a as well as C5a. We ruled this out as the depletion of FXII from plasma did not decrease complement inhibition by OSCS. In addition, C3 is an abundant protein in normal plasma with reference values of 0.67�C1.29 g/L, and thus unlikely to be consumed to levels that would impact C3 fixation.
convertases but was also dependent on FXII. The impact of GAGs on complement and the observed effect of OSCS on complement components C3 and C5 suggested further evaluation of OSCS and complement. Investigation of the OSCS interactions with complement components using surface plasmon resonance by Linhardt��s group suggested that OSCS can bind to the complement components with moderate to high affinity comparable to that of heparin. Therefore a difference in the impact of OSCS-contaminated versus uncontaminated heparin is not explained by differences in binding to complement components. The impact of OSCS on the functional complement activity was not performed yet, which became the aim of our study. In earlier work, the complement function has been tested in vitro using a variety of models such as the standard 50% hemolytic complement assay, the enzyme immunoassay and the liposome immunoassay. In this study, we used an established model of Foretinib natural antibody mediated bacterial lysis through the complement classical pathway. The murine monoclonal polyreactive antibody 2E4 can bind with bacteria E. coli BL21, fix complement, lyse bacteria and generate anaphylatoxin C5a. This is a relevant model to study the impact of OSCS, OSCS-contaminated heparin lots, un-contaminated heparin lots and other GAGs on the complement classical pathway. Using this biologically relevant model, antibody mediated complement-dependant bacterial lysis, we demonstrated that OSCS can inhibit the complement classical pathway as indicated by lower level of C3 fixation on bacteria as well as decreased bacterial lysis. Initially, we hypothesized that C3 consumption might explain the decreased fixation of C3b on the antibodytreated bacteria. This hypothesis was consistent with the FXIIdependent OSCS induction of C3a as well as C5a. We ruled this out as the depletion of FXII from plasma did not decrease complement inhibition by OSCS. In addition, C3 is an abundant protein in normal plasma with reference values of 0.67�C1.29 g/L, and thus unlikely to be consumed to levels that would impact C3 fixation.