Therefore presumably is subject to regulation by DUBs. Altering ubiquitination pathways may represent a way to modulate antibacterial autophagy and intracellular proliferation of pathogens. The ubiquitin system and the DUB enzymes themselves have become a new class of interesting therapeutic targets. Although no DUB inhibitors are yet in clinical trials, diverse inhibitors have already been described, including the USP14 inhibitor IU1 and inhibitors specific to USP7, USP2 and UCH-L3. In EX 527 addition, a small cell-permeable molecule, WP1130, also known as Desgrasyn, which selectively inhibits a subset of cellular DUBs, has been described recently as a potential anti-cancer therapeutic. This molecule causes depletion of monomeric ubiquitin molecules and accumulation of ubiquitinated proteins in cells. A previous study demonstrated that WP1130 directly inhibits in vitro activity of specific DUBs like USP9x, USP5, USP14 and UCH37, without affecting others, showing some degree of specificity. However, the fullspectrum of WP1130 DUB targets as well as its mechanism of action are still unknown. We previously found that WP1130 has anti-infective activity, reducing intracellular replication of some bacterial and viral pathogens. Replication of murine norovirus in a murine macrophage-like cell line, and other RNA viruses, were significantly reduced by WP1130. This antiviral activity was at least in part mediated by inhibition of USP14, a proteasome associated DUB that also controls induction of the unfolded protein response. Moreover, we recently showed that DUB inhibition by WP1130 increases ASP1517 HIF inhibitor killing of the gram-positive bacterium, Listeria monocytogenes, within macrophages. WP1130 treatment rapidly enhanced localization of the antimicrobial effector iNOS to the bacterial phagosome. These recent observations suggest that perturbation of protein ubiquination in host cells by small molecule DUB inhibitors may be an effective strategy to reduce infection that will cause minimal selective pressure on the pathogen itself. L. monocytogenes and noroviruses, along with Salmonella and Toxoplasma, are among the leading causes of food and waterborne diarrheal diseases worldwide. In the US alone, food-borne pathogens are estimated to cause 9.4 million infections and around 1,350 deaths annually, and 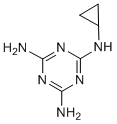 are associated with substantial healthcare costs. However, there are limited vaccines or antimicrobial drugs available to prevent or treat these infections. As the DUB inhibitor WP1130 showed activity against two of these food-borne pathogens, exploiting the ubiquitin system for the development of a new class of broad-spectrum therapeutics to treat these infections is appealing. However, therapeutic use of WP1130 itself is limited due to its low solubility and poor bioavailability in animals. Here we use L. monocytogenes as a model pathogen to screen a small library of WP1130-derivative molecules for antiinfective efficacy in macrophages, a major reservoir for many intracellular pathogens, with the overall goal of finding derivatives with better solubility and anti-infective activity. Several authors recently reported that the incretin system induces an inflammatory and pro-lipolytic response via the PKA NF-kB – IL-1 pathway and impairs insulin sensitivity and glucose uptake in human adipocytes. One of the key mechanisms in the pathogenesis of diabetes-related vascular dysfunction is oxidative stress. Oxidative stress is attributable to excessive production of reactive oxygen species and inflammatory markers by tumor necrosis factor-alpha, macrophage chemotactic protein-1 and other markers. The inflammatory response was reported to downregulate eNOS expression and upregulate iNOS expression in rodents and increase NADH oxidase activity and vascular remodeling.
are associated with substantial healthcare costs. However, there are limited vaccines or antimicrobial drugs available to prevent or treat these infections. As the DUB inhibitor WP1130 showed activity against two of these food-borne pathogens, exploiting the ubiquitin system for the development of a new class of broad-spectrum therapeutics to treat these infections is appealing. However, therapeutic use of WP1130 itself is limited due to its low solubility and poor bioavailability in animals. Here we use L. monocytogenes as a model pathogen to screen a small library of WP1130-derivative molecules for antiinfective efficacy in macrophages, a major reservoir for many intracellular pathogens, with the overall goal of finding derivatives with better solubility and anti-infective activity. Several authors recently reported that the incretin system induces an inflammatory and pro-lipolytic response via the PKA NF-kB – IL-1 pathway and impairs insulin sensitivity and glucose uptake in human adipocytes. One of the key mechanisms in the pathogenesis of diabetes-related vascular dysfunction is oxidative stress. Oxidative stress is attributable to excessive production of reactive oxygen species and inflammatory markers by tumor necrosis factor-alpha, macrophage chemotactic protein-1 and other markers. The inflammatory response was reported to downregulate eNOS expression and upregulate iNOS expression in rodents and increase NADH oxidase activity and vascular remodeling.
Category: clinically Small Molecule
Among mesenchymal cell types that dynamically populate both developing and injured tissues are cells of the innate immune system
Hence, high numbers of macrophages colonize virtually all epithelial tissues early in embryogenesis, and key trophic effects of this immune cell subset have been inferred by the severely impaired growth of epithelial organs displayed by animal models deficient in macrophages or macrophage-dependent functions. Recruitment of myeloid cell populations from the bone marrow to the periphery continues to be essential in adulthood for the maintenance of tissue integrity, since, in their absence, tissue repair and regenerative events following injury are critically blunted. To date, experimental evidence indicate that macrophages may primarily influence the growth and/or regeneration of WY 14643 epithelial organs indirectly, i.e. by supporting functions such as clearance of dying cells, angiogenesis and remodeling of extracellular matrices. Whether macrophages can directly dictate select developmental options in epithelia remains presently unclear. During pancreatic development, at E14.5–15.5 gestational age, epithelial progenitors emerge from a rudimentary ductal tree through a regulated sequence of events that includes withdrawal from the cell cycle, delamination into the surrounding mesenchyme and differentiation into endocrine or exocrine cell types. As such, while providing a pool of progenitors competent to execute specific developmental steps and make divergent lineage choices, the E14.5/E15.5 pancreas represents a valuable model to study how such epithelial programs might be impacted on by other exogenous cellular cues. In this regard, in vivo and in vitro studies have provided evidence that,WZ4002 over-imposed to a hierarchy of transcription factors expressed by the epithelium, interactions of the epithelium with the pancreatic mesenchyme govern the balance between the exocrine and the endocrine developmental fate of progenitors and are required for the growth of the pancreatic epithelial compartment as a whole. At present, few studies have reported the presence of tissue macrophages within the pancreatic mesenchyme and noted reduced growth of endocrine cells in their absence. However, the possible role of macrophages as regulators of select developmental events in the pancreatic epithelium remains unknown. A corollary to this question is whether diverse states of activation of tissue macrophages differentially affect pancreatic developmental programs. Indeed, macrophages resident within tissues may adopt a spectrum of functional states. At the extreme of this spectrum are classically and alternatively activated macrophages. M1 phenotypes are acquired by macrophages upon encounter with pathogens, and lead to the production of high levels of pro-inflammatory mediators and reactive nitrogen intermediates that contribute to pathogen clearance. Conversely, ‘‘alternative’’ or M2 activation states are characterized by the production of lower levels of pro-inflammatory cytokines, synthesis of decoy anti-inflammatory receptors, little or no nitrogen derivatives, as well as production of mediators of tissue remodeling. As such, M2 macrophages have immuneregulatory functions that dampen inflammation and promote repair during wound healing.
Resolution limit in community detection methods is also manifested in the size and statistical significance of modules
Note that while more than 45% of extracted genes were retained under the most stringent B-score cutoff used, such robustness against statistical significance cutoffs was not observed for other algorithms such as MCODE and modularitybased community detection. Even at a less stringent B-score cutoff of 0:05, the MCODE and modularity-based modules would generally suffer from a loss of over 50% and 95% of identified genes, respectively. Therefore, we did not include the B-score significance measure for the MCODE modules in all comparative analyses. Using the Rembrandt grade II glioma data as an example, the largest module identified by the community detection method as of, consisting of 1,372 genes out of a total of 3,888, was deemed statistically non-significant under the B-score scheme. A careful inspection of this large module showed that three of the statistically significant DiME modules, with sizes of 212, 39 and 42 genes respectively, are contained or almost contained within it. It also has significant overlaps with several other non-significant DiME modules. In comparison, three MCODE modules are contained within the above mentioned large module,Rapamycin with sizes of 77, 18 and 13 genes respectively. Such an observation suggests that community detection is not appropriate for disease module identification in large biological networks, since it generates huge modules with large numbers of genes which add difficulties to validation and interpretation. An analysis of the variability of module identification results show that core modular structure of the Rembrandt coexpression networks used in the case study is well conserved under varying network construction parameters. Such conservation is consistent with the concept of ‘‘module core’’ described by the original authors of module extraction. It is worth pointing out, however, that the less conserved modules do not necessarily bear little functional significance in the network, as their fluctuations may be due to the noise in the biological data itself, rather than in the module identification algorithm. The construction of a highly robust network per se is still a highly active area of research and is not the main focus of this paper. The module connectivity networks for SAR131675 grade II glioma and GBM samples provide a high-level yet insightful understanding of brain tumour progression and the associated rewiring of cellular machinery. A common expression signature of both tumour grades is down-regulation of nervous system development and normal neuronal functions and upregulation of cell cycle related progresses, light green and red nodes). Such concomitant alterations in transcriptome are consistent with a malignant phenotype – cells that are becoming less differentiated and are proliferating more. The coordination between the two types of functional processes is remarkably strengthened in GBM compared with grade II glioma samples, a possible consequence of the significant increase in the transcription factors AR and ETS1 shared by the two processes in both grades. Core components of the two processes are also conserved across microarrays, as is shown by the expression levels of modules 2, 3, and 6 in Figure 7. Also of pathological significance is the significant increase in the activity of the angiogenesis-related module in GBM.
The C-terminal PTH-unrelated region of PTHrP containing the osteostatin epitope may also contribute to its osteogenic actions
These results suggest that Asian males should be offered testing for defects in ACE I/D polymorphisms, especially if they are hypertensive. We suggest that physicians should provide specific protection to D-allele carriers, for example by administering ACE inhibitors to hypertensive patients. The insulin-like growth factor system, formed by insulinlike peptides, their receptors and binding proteins, plays a central role in the regulation of cell growth and differentiation. Human homozygous loss-of-function IGF1 gene mutations cause intrauterine and postnatal growth failure and severe sensorineural deafness. Treatment with recombinant human IGF-I has been shown to improve short stature in patients with severe IGF-I deficiency, supporting the key role of IGF-I in skeletal development. Moreover, decrease in IGF-I production and/or activity has been suggested to contribute to age-related osteopenia and low bone formation. Mice with a homozygous Igf1 gene deletion display a 30% size reduction and an aberrant bone phenotype with shortened femoral length and reduction in cortical bone size, and also sensorial impairment, MG132 as compared to wild type littermates. These bone changes are related to a decrease in both bone formation and bone resorption with a low number of osteoblasts and osteoclasts, and also a reduced capacity for osteoblastogenesis and osteoclastogenesis in the bone marrow of Igf1-null mice. Thus, the observation of an increased trabecular bone volume in the proximal tibia of these mice was suggested to be a consequence of the IGF-I effect on osteoclast formation and/or activity at this skeletal site, which is absent in Igf1-null mice. Mice with a homozygous deletion of the gene encoding the IGF-I high affinity receptor show a delayed ossification in the cranial and facial bones, inner ear alterations and die shortly after birth. Furthermore, partial deletion of the Igf1r gene causes postnatal growth retardation in humans. IGF1R activation recruits insulin receptor substrates. Mice with homozygous deletion or spontaneous mutation in Irs1 show sensory alterations,MK-1775 severe bone growth impairment and low-bone turnover osteopenia. In addition, gain-of-function mouse mutants of IGF-I binding proteins that reduce IGF-bioavailability also consistently show a low cortical and trabecular bone mineral density and alterations in bone formation rates. The bulk of current studies performed in rodents support the notion that the IGF system plays a paramount role in the bone anabolic actions of PTH. Thus, neither Igf1-null nor Igf1r-null mice show the bone anabolic response triggered by transient administration of PTH in normal mice. IGF1R in mature osteoblasts seems to be a critical PTH target for its skeletal actions. Cells of the osteoblastic lineage are a rich source of PTHrelated protein, an important modulator of bone development and bone remodelling. PTH and PTHrP interact with the same PTH type 1 receptor in osteoblasts. Similarly to PTH, intermittent administration of the N-terminal PTHrP fragment induces bone anabolic features in mice and humans.
The information required for establishing an interaction with another protein is already present in the tridimensional structure
In any case, one cannot ignore the fact that the characteristics of the protein environment also can play an important role by being able to modify protein structures and, consequently, interfaces. Additionally, it is also clear that if a protein is interacting with two or more different partners, different interfaces may be formed for each partner. A careful literature review will quickly confirm that although there are several recently published studies regarding the characteristics that could determine differences between interface-forming residues and free surface residues, there is no general agreement about exactly how proteins associate with each other and which descriptors of their characteristics are suitable for elucidating this mechanism. Also, by comparing the interface area against the rest of the free surface is a common procedure during attempts to characterize the main differences between those two classes. This type of comparison has been described in recent studies, including those cited above Fulvestrant. A variety of models and descriptors were explored to build protein-protein interface classifiers. Promate and PINUP used linear scoring functions, while PPI-Pred used a support vector machine approach, SPPIDER and cons-PPISP used a neural network model, and Meta-PPISP combined the results of cons-PPISP, Promate and PINUP as a meta-predictor. In contrast to the method proposed in this study, the six mentioned models make use of amino acid sequence conservation and propensity. How important is this difference among StingLDA and other mentioned algorithms could only be accessed adequately if proper analysis is done on how often the conservation property could not be used in known protein universe. It is known that structural genome projects used high-throughput techniques to produce and then deposit in the PDB thousands of new structures. For instance, half of the protein structures solved during the year of 2005 came from structural genome initiatives, including structures of the so-called orphan proteins. Orphan proteins are organism-specific proteins, i.e. GANT61, they have no homologue protein in other lineages. Estimates are that up to one third of the genes/ proteins from whole known genomes accounts for orphan proteins. Ekman et al. show, using the structural classification of protein, that up to 25% of the known non-redundant protein structures from bacteria are from orphan proteins or from proteins having an orphan domain. Also, up to 21% of known protein structures in Eukarya kingdom and 24% in Archaea kingdom follow the same trend. Operating in such scenario where limitations imposed by orphan protein existence restricts the use of aforementioned algorithms dependent on conservation parameter for predicting interface residues, would clearly lead to unreliable results. Therefore, the strong demand is created for the development of more general approaches for IFR prediction which would have similar performance to conservation dependent algorithms, yet without the use of evolutionary-related attributes for prediction. The Sting-LDA was produced having in mind this demand as well. We report results on the classification of the 20 naturally occurring amino acids into two distinct classes: IFR and FSR, by using several amino acid descriptors from the BlueStar STING database. BlueStar STING has been used previously for predicting enzyme class, protein-ligand analysis, protein mutant analysis, and protein-protein interaction pattern analysis, mostly because BlueStar STING offers easy access to a very rich repository of protein characteristics.